Schwindel ist ein meist unangenehmes Empfinden, das durch eine Scheinbewegung der Umwelt oder eine scheinbare Selbstbewegung sowie durch eine Störung des Schwerkraftgefühls und der Raumorientierung hervorgerufen wird.

Das Symptom Schwindel zählt generell zu den häufigsten beklagten Beschwerden. 20-30% aller Erwachsenen und die zum Arzt gehen leiden unter Schwindel, im Alter ist Schwindel das häufigste Symptom überhaupt. In einer deutschen Bevölkerungsstichprobe berichteten 29% von 8318 Teilnehmern der Befragung über mäßig bis schwer ausgeprägte Schwindelepisoden in ihrem bisherigen Leben, ein Viertel davon litt unter Drehschwindel. Andere Studien gehen sogar davon aus, dass durchschnittlich jeder Dritte einmal im Jahr einen Schwindelanfall erleidet, bei den über 70-Jährigen sogar jeder Zweite. Bei den meisten Betroffenen tritt der Schwindel wiederkehrend auf, und verursacht eine erhebliche Beeinträchtigung. Entsprechend vielfältig sind die sich oft dahinter verbergenden Krankheiten und Befindlichkeitsstörungen. Es handelt sich um ein vieldeutiges Symptom. Schwindel wird oft als bedrohlich empfunden. Menschen, die unter heftigem Schwindel leiden, fühlen sich schwer krank. Wenn Menschen von Schwindel sprechen, meinen sie oft ganz unterschiedliche Empfindungen. Allein die Unterscheidung zwischen Übelkeit und Schwindel ist oft schwierig. Dies liegt nicht nur daran, dass jeder etwas anderes meint, wenn er von Schwindel spricht. Die erste Schwierigkeit bei der Schwindeldiagnose ist deshalb zu verstehen was der Betroffene unter Schwindel versteht. Ist der Arzt einseitig festgelegt fragt er nicht selten, das was er hören will in den Patienten hinein. Ausredenlassen, und erst dann gezieltes Nachfragen erleichtert die Diagnosefindung. Die erste Frage bei der Untersuchung ist also, was der Patient unter Schwindel versteht: Drehschwindel mit empfundenem Drehen der Umgebung?, Bewegungsunsicherheit?, Gangunsicherheit? Bewusstseinsstörung?, Übelkeit?, Aufmerksamkeitsstörungen und –schwankungen?, Antriebsstörung?, Benommenheit, Leeregefühl im Kopf?, Kopfdruck?, Gang- und Standunsicherheit? Sehstörungen?, Gefühl der Angst/Unsicherheit?, Derealisation oder Depersonalisation- das Gefühl als ob die Umgebung oder die Person verändert sind? Wahrnehmungsstörungen? Schwankschwindel? Tritt der Schwindel anfallsweise auf, ist es ein Dauerschwindel, wie lange dauern die Anfälle, sind sie durch Lageänderungen oder Kopfbewegungen ausgelöst, was gibt es für Begleitsymptome? Die Antwort auf diese Fragen ist wegweisend für die Diagnose und ermöglich auch ohne technische Untersuchung in vielen Fällen bereits die Diagnose.
Differentialdiagnose von Schwindel
| Diagnose | Symptome, diagnostische Kriterien |
| gutartiger Lagerungsschwindel | Kurze Dreh-Schwindelepisoden bei Lagewechsel mit einer positiven Antwort auf das Hallpike Manöver. Mit dem Alter zunehmend häufig, das häufigste Drehschwindelsyndrom. 0.5% bei den 18-39-jährigen 3,4% pro Jahr bei den über 60-jährigen |
| Neuronitis vestibularis | Akuter Dauerdrehschwindel. Zweit häufigstes Drehschwindelsyndrom. Meist zwischen dem 30.und 50. Lebensjahr, häufiger gefolgt von psychogenem Schwindel (etwa ein Drittel der Patienten) |
| Morbus Menière | Dreh-Schwindelepisoden, Tinnitus, Hörminderung, 3-11% der Patienten von speziellen Schwindelambulanzen, in der Allgemeinbevölkerung selten und eher zu häufig diagnostiziert. |
| Perilymphatische Fistel | Abnorme Verbindung zwischen Mittelohr und Innenohr und hierdurch Leck der Perilyphflüssigkeit mit Eindringen von Luft in die Kochlea. Kann mit dem Morbus Menière verwechselt werden. Impulsartige Druckänderung beim Husten, Niesen, Pressen oder Heben im Bereich des Mittelohres oder des Liquorraumes führen hier zu Drehschwindel mit Nystagmus. Akutes Auftreten nach Mittelohroperation, Flugzeuglandung, Vasalvamanöver…Die Diagnose wird durch chirurgische Exploration gestellt bzw. bestätigt. |
| Cervicale Spondylose | Symptome bei Kopf und Halsbewegung mit eindeutig eingeschränkter Nackenbeweglichkeit. Diese Ursache bzw. Diagnose ist wissenschaftlich sehr umstritten, auch viele andere Schwindelursachen haben diese Symptome. Die HWS als Ursache wird mit Sicherheit zu häufig diagnostiziert. (Vorsicht kann auch sekundär zu Angst sein, Verspannung bei Angst begleitet fast jede Schwindelerkrankung). Es Experimente mit Lokalanästhesie von Gelenksrezeptoren der HWS, die bewiesen haben, dass durch Reizung und Ausschaltung von Rezeptoren (anders ist eine cervikale Genese nicht denkbar) keines der häufig geschilderten Schwindelsyndrome hervorgerufen werden kann (laut Brandt und Diener), allenfalls eine rasch innerhalb von wenigen Tagen kompensierte isolierte Gangunsicherheit. Dass Schwindel von der HWS ausgehen könnte ist insgesamt sehr umstritten. Die Annahme eines cervikalen Schwindels beruht möglicherweise auf Fehlinterpretationen von Tierversuchen. Siehe auch: Roy J.E. and Cullen K.E., Passive Activation of Neck Proprioceptive Inputs Does Not Influence the Discharge Patterns of Vestibular Nuclei Neurons. New York Academy of Sciences, 942:486-9, 2001. T Brandt and A M Bronstein NOSOLOGICAL ENTITIES?: Cervical vertigo J Neurol Neurosurg Psychiatry 2001; 71: 8-12.[Full text] Richmond FJR, Bakker DA. Anatomical organization and sensory receptor content of soft tissues surrounding upper cervical vertebrae in the cat. J Neurophysiol 1982;48:49-61 Tadashi Nakamura, Brain, Vol. 118, No. 5, 1157-1168, 1995 |
| Orthostatische Hypotension | Auch den meisten gesunden Menschen ist es schon einmal passiert, dass nachdem sie lange in der Sonne gelegen haben, nach der ersten Zigarette, übermüdet waren oder gerade von einer mehr oder weniger langen Bettlägerigkeit genesen sind, beim schnellen Aufrichten ein Leeregefühl im Kopf, ein Schwarzwerden vor den Augen empfunden haben und sich schnell wieder hinsetzen mussten um nicht hinzufallen. Was bei seltenem Vorkommen und wenn die akute vorübergehende Ursache bekannt ist, harmlos und leicht verkraftbar ist, kann sehr lästig sein, wenn es ständig auftritt. Erst dann spricht man von einer Orthostatischen Hypotension, einem krankhaften Blutdruckabfall beim Aufrichten. Es handelt sich um eine fehlende adäquate Blutdruckreaktion beim Aufstehen aus sitzender oder liegender Position. Häufigkeit nimmt mit dem Alter zu. Bei 20% der über 65 jährigen vorhanden, aber nur jeder 9. davon hat Symptome. Fallen um 20 mm Hg für den systolischen Druck oder 10mm Hg für den diastolischen Blutdruck in einer Minute mit begleitenden Symptomen der Minderdurchblutung des Gehirns( nach Lagewechsel Liegen zu Stehen) Näheres zum Schwindel durch zu schnelles Aufrichten. |
| Zerebrale Durchblutungsstörungen | Diffuse Durchblutungsstörungen machen einen diffusen Schwindel mit Leeregefühl im Kopf, bei schwerer wiegenden DBS mit Schädigung der weißen Substanz kommen Gangstörung (marche a petits pas) Reflexsteigerung Tonussteigerung, Gangstörung mit oder ohne Paresen, Verlangsamung, Inkontinenz etc. dazu. Sonderfälle sind Stealsyndrome, Gefäßstenosen, Diffuse DBS sind auch im Alter eher seltene Ursache von Schwindel, werden aber häufig diagnostiziert. |
| Schlaganfall | In Folge von Hirnstamm und Kleinhirninfarkten kann es zu einer akuten Drehschwindelsymptomatik kommen. Meist sind dann andere neurologische Symptome mit vorhanden, wie Ataxie, Hemiparese, Hirnstammzeichen mit Hirnnervenausfällen etc. Siehe z.B. unter Medulla-oblongata-Syndrome. In Notfallambulanzen haben etwa 3% der Schwindelpatienten einen Schlaganfall. Selten kann bei einem Kleinhirninfarkt ein isolierter Drehschwindel vorliegen (die sich dann über Tage bessert), meist besteht zusätzlich eine Ataxie. Bei unkorrekter Untersuchung sind Verwechslungen mit einer Vestibularisneuritis möglich, da auch hier eine akuter Dauerdrehschwindel bestehen kann. Bei Dissektionen der Vertebralarterien oder auch allgemein beim Kleinhirninfarkt können auch Hinterkopfschmerzen auftreten, die zur Verwechslung mit Migräne Anlass geben können. |
| Labyrinthblutung | akuter Hörverlust und Drehschwindel. Selten kommt bei Einnahme gerinnungshemmernder Medikamente, Kokainmissbrauch, Leukämien und Sichelzellanämie vor. Diagnose im MRT |
| Ataxien | Bewegungsstörung vorrangig, chronischer Verlauf. Bei der seltenen episodische Ataxie Typ II episodische Schwindelattacken, Okulomotorikstörungen im Intervall. Die episodische Ataxie Typ II ist mit Azetazolamid und dem Kaliumkanalblocker 4-Aminopyridin prophylaktisch behandelbar. |
| sensible Ataxie | Gang und Standunsicherheit (wird häufig als Schwindel empfunden), die im Dunkeln bei fehlender visueller Orientierung zunimmt und durch fehlende propriozeptive Information verursacht wird. Verlust der Tiefensensibilität, des Lagesinns und Bewegungsempfindens, hauptsächlich bei Polyneuropathie, aber auch bei Rückenmarksschädigungen. |
| Angst | Angstsymptome erfragbar, manchmal Reproduktion der Symptome bei Hyperventilation. Häufige Ursache eines diffusen Schwindels. Nicht selten auch Folge organischer Schwindelerkrankungen. Wenn die Angst vor dem Schwindel zum ängstlichen Insichhineinhören wird, wird aus der Angst vor dem Schwindel schnell die Angst vor der Angst. Wegen dem hohen Risiko der Chronifizierung und dem dann erheblichen Ausmaß der Behinderung sollte dann möglichst bald fachärztliche Hilfe gesucht werden. |
| Sehstörung | Sehschärfe reduziert auf weniger als 6/9 auf beiden Augen. Augenmuskellähmung, Myasthenie, Seltene Ursache |
| Allgemeinerkrankungen | Diffuser Schwindel „Leeregefühl im Kopf“, Schwarzwerden vor den Augen“.. Blutarmut (Anämie, z.B.: nach Blutverlust Vitamin B12 oder Eisenmangel, heftige Monatsblutungen…), Infekte, Schwäche nach Bettlägerigkeit, Herzrhythmusstörungen, Unterzuckerung (Fast nur bei Diabetes oder Alkoholikern nach heftigem Trinken), Atemnot. Auch die Behandlung kann den Schwindel auslösen, siehe unten unter Medikamente. |
| Sucht | Schlafmittel, Beruhigungsmittel, Alkohol, Drogen |
| Nach vielen Mittelohrentzündungen | Cholesteatom? |
| Bewegungsschwindel oder die Seekrankheit | typische Vorgeschichte |
| Vestibuläre Epilepsie | sehr selten, typische Anamnese. Diffuser Schwindel kann aber mit Absencen oder komplex fokalen Anfällen verwechselt werden. |
| Bilaterale Vestibulopathie | Stand- und Gangunsicherheit bei Dunkelheit und auf unebenem Grund, bei raschen Kopfbewegungen Gehen, Laufen oder Fahren können die Patienten nicht mehr scharf sehen, Straßenschilder oder entgegenkommende Menschen werden nicht sicher erkannt. Ursachen der beidseitigen Vestibularisschädigung können ganz unterschiedlich sein beispielsweise Medikamenten wie Aminoglykoside oder Autoimmunerkrankungen. Die Behandlung richtet sich nach der Ursache. Gang- und Gleichgewichtstraining ist aber, wie bei den meisten Schwindelerkrankungen in jedem Fall sinnvoll. |
| paroxysmaler Schwindel | kurzdauernde Drehschwindelattacken, seltener auch Schwankschwindelattacken, die häufig durch Kopfbewegungen ausgelöst werden aber auch in Ruhe auftreten können und auf eine Behandlung mit Antiepileptika meist ansprechen. Ausgelöst durch pathologischen Gefäß-Nervkontakt am VIII. Hirnnerv (meist Schlinge der A. cerebelli inferior anterior (AICA)). Entsprechend kann in hartnäckigen Fällen eine mikrovaskuläre Dekompression heilend sein. Diagnostische Kriterien: 1) kurze Dreh- oder Schwankschwindelattacken (Dauer: Sekunden bis Minuten), 2. häufig Auslösung der Attacken durch bestimmte Kopfpositionen und Beeinflussung der Anfallsdauer durch Veränderung dieser Position, 3. permanent vorhandene oder während der Attacke auftretende otologische Zusatzsymptome (Hypakusis und Tinnitus), 4. neurophysiologischer Nachweis der auditorischen und vestibulären Störung, 5. therapeutische Wirkung von Carbamazepin oder anderen Antiepileptika 6. Ausschluss einer anderen zentralen Schwindelursache. |
| Synkopen oder Ohnmachten | muss von der Angst in Ohnmacht zu fallen unterschieden werden |
| Basilarismigräne. | Häufig Familienvorgeschichte mit Migräne. Häufig mit anderen Aurasymptomen wie visuellen Symptomen in beiden temporalen und/oder nasalen Gesichtsfeldern beider Augen Doppelbilder, Dysarthrie, Tinnitus, Hörminderung, Ataxie, beidseitige Körpermissempfindungen, beidseitige Lähmungen, selten auch Bewusstseinsverlust. Der Schwindel der Basilarismigräne ist meist gefolgt von Kopfschmerzen. Diese halten meist 4–72 Stunden an, sind oft einseitig, pulsierend, mäßig bis schwere, nehmen bei körperlicher Aktivität zu, nehmen oft bei Lärm oder grellem Licht zu. (Nach manchen Angaben sollen aber 30% der Basilarismigräneattacken ohne Kopfschmerzen sein). Dauer des Schwindels über manchmal Sekunden manchmal Stunden, reversible neurologische Defizite, Zwischen den Attacken häufig Auffälligkeiten mit Störung der zentralen Okulomotorik in Form einer sakkadierten Blickfolge, Blickrichtungsnystagmus, Spontannystagmus oder Lagerungsnystagmus. Es handelt sich um eine Ausschlussdiagnose. Ein Migräne-Schwindel kann diagnostiziert werden, wenn ein Migräne-Syndrom nach IHS-Kriterien und ein episodischer vestibulärer Schwindel bestehen und beide während mindestens zweier Attacken gemeinsam aufgetreten sind. Die übliche Migräneprophylaxe ist wirksam. |
| Migräne | Schwindel ist bei Migräne häufig (etwa 1% der Bevölkerung), die überwiegende Basilarismigräne ist aber selten. Der Schwindel im Rahmen einer allgemeinen Migräneattacke kann von Sekunden bis zu Tagen dauern, im Rahmen der Basilarismigräne handelt es sich um 5-60min dauernde Aurasymptome. Auch beim Migräneschwindel findet man häufig einen pathologischen Nystagmus. Von einem definiten Migräneschwindel geht man aus, wenn: (1) Episodisch vestibuläre Symptoms von mäßigem Schweregrad auftreten (a) Drehschwindel (b) Lagerungsschwindel (c) Beschwerdezunahme bei Kopfbewegung (2) Migräne nach den Kriterien der International Headache Society (3) eines oder mehr Symptome während mindestens 2 Schwindelattacken (a) migränöser Kopfschmerz (b) Kopfschmerz (c) Lichtscheu (d) Geräuschempfindlichkeit (e) Migräneaura (4) Andere Diagnosen wurden durch adäquate Untersuchung ausgeschlossen Von einem wahrscheinliche Migräneschwindel geht man aus, wenn: (1) Episodisch vestibuläre Symptoms von mäßigem Schweregrad auftreten (a) Drehschwindel (b) Lagerungsschwindel (c) Beschwerdezunahme bei Kopfbewegung (2) eines der folgenden Symptome (a) Migräne nach den Kriterien der International Headache Society (b) Migränesymptome während des Schwindels (c) migränetypische Auslöser des Schwindels (z.N. spezielle Nahrungsmittel, Schlafmangel…) (d) Ansprechen auf Migränebehandlung (3) Andere Diagnosen wurden durch adäquate Untersuchung ausgeschlossen (Neuhauser H, Leopold M, von Brevern M, et al. The interrelations of migraine, vertigo, and migrainous vertigo. Neurology 2001; 56:436-441). Vlasta Vukovic et al., Prevalence of Vertigo, Dizziness, and Migrainous Vertigo in Patients With Migraine Headache 2007;47:1427-1435 Abstract |
| gutartiger paroxysmaler Schwindel des Kindesalters | plötzlich einsetzende Drehschwindelattacken mit Blässe, Nystagmus, Erbrechen und Angstgefühl charakterisiert. Zwischen den Attacken ist der neurologische Befund unauffällig. Kleinkinder bis zum 8. Lebensjahr, Migräneäquivalent. Ähnlich auch zyklisches Erbrechen mit gastrointestinalen Beschwerden ohne fassbares organisches Substrat mit Blässe und Lethargie bei Kindern. Häufigster Drehschwindel bei Kindern |
| verschiedene Medikamente | meist orthostatischer Schwindel oder diffuser Schwindel. Alkohol, Drogen, Blutdruckmedikamente (besonders bei Überdosierung oder zu Beginn der Behandlung), Antiepileptika (besonders Carbamazepin dann aber Drehschwindel am Anfang der Behandlung und bei Überdosierung), Schmerzmittel und Entzündungshemmer, Schlaf- und Beruhigungsmittel, Muskelrelaxantien, Medikamente gegen Übelkeit, Antidepressiva, Anticholinergika, Dopaminagonisten, Lokalanästhetika Antibiotika, Tuberkulostatika, Antimykotika, Anthelminthika, Betablocker, Vasodilatatoren, -konstriktoren, Diuretika, Spasmolytika, Antiallergika, Röntgenkontrastmittel, Beachten Sie, wenn Sie mehrere Medikamente nehmen, kann die Wechselwirkung der Medikamente auslösend sein. Bei Schwindel bei Menschen, die viele Medikamente nehmen, kann es sinnvoller sein in Absprache mit dem Arzt einzelne Medikamente abzusetzen, als neue dazu zugeben. |
| Andere | Z.B.: M. Parkinson periphere Polyneuropathie Multiple Sklerose, Infektionen. Besondere Vorsicht ist bei der Behandlung von Schwindel bei M. Parkinson, oft werden gegen Schwindel Neuroleptika eingesetzt, die bei M. Parkinson, zu massiver Verschlimmerung der Symptome führen können. |
| Oft keine Diagnose möglich | – |
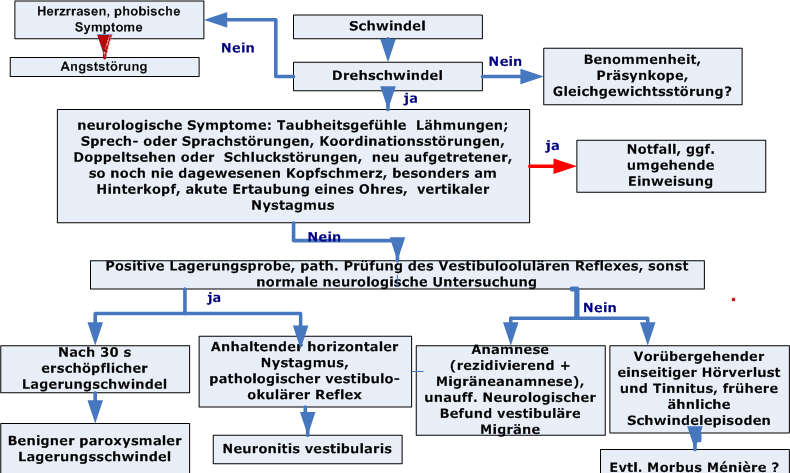
Diagnostischer Algorithmus
Häufigkeiten verschiedener Schwindel-Syndrome
| Tabelle: Häufigkeiten verschiedener Schwindel-Syndrome bei ambulanten Patienten einer Spezialambulanz. Statistische Daten der Schwindelambulanz der Neurologischen Klinik des Klinikums Großhadern in München Dieterich 1996). | ||
| Summe 1989-1995 | ||
| Diagnose | n | % |
| gutartiger anfallsweise auftretender Lagerungsschwindel BPPV | 258 | 18,8 |
| Phobischer Schwankschwindel (Angstsymptom), | 196 | 14,3 |
| Zentral-vestibulärer Schwindel | 185 | 13,5 |
| Morbus Menière | 101 | 7,3 |
| Basiläre Migräne | 83 | 6,0 |
| Neuronitis vestibularis | 67 | 4,9 |
| Psychogener Schwindel (ohne phobischen Schwankschwindel) | 41 | 3,0 |
| Pathologischer Gefäß-Nerv-Kontakt (paroxysmaler Schwindel) Vestibularisparoxysmie | 24 | 1,8 |
| Bilaterale Vestibulopathie | 31 | 2,3 |
| Otolithenschwindel | 3 | 0,2 |
| Labyrinthfistel | 3 | 0,2 |
| Schwindel unklarer Ätiologie | 64 | 4,7 |
| Andere zentral-vestibuläre Syndrome ohne Schwindel | 186 | 13,8 |
| Andere Erkrankungen ohne Schwindel | 100 | 13,8 |
| Gesamt | 1370 |
