autosomal-rezessive Kupferspeicherkrankheit, durch Mutationen am Genort der kupfertransportierenden ATP-ase 7B (13q 14.3). Tritt in der Allgemeinbevölkerung mit einer Häufigkeit von 1/30 000- 1/40000 auf. 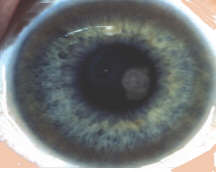 Ein gestörter Kupfertransport durch die Zellmembranen führt zur Akkumulation von Kupfer in verschiedenen Organen, wobei die Leber und das Gehirn am stärksten betroffen sind. Die Kupferablagerung in der Leber tritt früh im Verlauf auf und führt zu zunehmender Leberfunktionsstörung mit portaler Hypertension. Überschüssige Kupferablagerungen im Hirn führt zu Nervenschäden, motorischen Störungen, kognitiven Beeinträchtigungen und Verhaltensstörungen. Am Beginn tritt meist eine Lebersymptomatik (meist schon bei Kindern) auf, in der Pubertät und bis zum 30. Lebensjahr kommen dann neurologische und psychiatrische Symptome dazu. Neurologische Symptome: Zittern, Sprachstörungen, Schwierigkeiten beim Gehen und Schreiben, vermehrter Speichelfluss, Versteifung und Fehlstellung der Gliedmassen; psychiatrische Auffälligkeiten reichen von Psychose, verminderter geistiger Leistungsfähigkeit bis zu schweren Verhaltensstörungen. Die Diagnosesicherung erfolgt laborchemisch. Bei verdächtigen Symptomen wird eine Familienanamnese erhoben, an den Augen zeigt sich häufig ein Kayser-Fleischer Kornealring bei der Spaltlampenuntersuchung, niedriges Serumkupfer und Ceruloplasmin bei erhöhter 24 Stunden Ausscheidung von Kupfer. Bei Patienten mit neurologischen Symptomen findet man immer auch kernpsintomographischen Auffälligkeiten im Gehirn, in der Regel mit einer Atrophie, meist auch mit Signalauffälligkeiten in verschiedensten Hirnregionen. (Neuroradiology (2006) 48: 613–621) Mit Chelatbildnern kann man die Erkrankung erfolgreich behandeln, manchmal ist eine Lebertransplantation erforderlich. Bei unbehandelten Patienten ist der Verlauf progredient und führt zum Tod. Regelmäßige Behandlung und Überwachung führt meist zu erheblicher Besserung. Die Behandlung ist allerdings lebenslang erforderlich. Unter Behandlung sind Lebenserwartung und -qualität meistens weitgehend normal. Eingesetzt werden D-Penicillamin, Trientine, Zink und Tetrathiomolybdat. Die Behandlung muss ohne Unterbrechung lebenslang fortgeführt werden, auch während einer eventuellen Schwangerschaft. Zu Todesfälle oder schlechtem Verlauf kommt meist bei fehlender Behandlung oder unzureichender Mitarbeit bei der Behandlung.
Ein gestörter Kupfertransport durch die Zellmembranen führt zur Akkumulation von Kupfer in verschiedenen Organen, wobei die Leber und das Gehirn am stärksten betroffen sind. Die Kupferablagerung in der Leber tritt früh im Verlauf auf und führt zu zunehmender Leberfunktionsstörung mit portaler Hypertension. Überschüssige Kupferablagerungen im Hirn führt zu Nervenschäden, motorischen Störungen, kognitiven Beeinträchtigungen und Verhaltensstörungen. Am Beginn tritt meist eine Lebersymptomatik (meist schon bei Kindern) auf, in der Pubertät und bis zum 30. Lebensjahr kommen dann neurologische und psychiatrische Symptome dazu. Neurologische Symptome: Zittern, Sprachstörungen, Schwierigkeiten beim Gehen und Schreiben, vermehrter Speichelfluss, Versteifung und Fehlstellung der Gliedmassen; psychiatrische Auffälligkeiten reichen von Psychose, verminderter geistiger Leistungsfähigkeit bis zu schweren Verhaltensstörungen. Die Diagnosesicherung erfolgt laborchemisch. Bei verdächtigen Symptomen wird eine Familienanamnese erhoben, an den Augen zeigt sich häufig ein Kayser-Fleischer Kornealring bei der Spaltlampenuntersuchung, niedriges Serumkupfer und Ceruloplasmin bei erhöhter 24 Stunden Ausscheidung von Kupfer. Bei Patienten mit neurologischen Symptomen findet man immer auch kernpsintomographischen Auffälligkeiten im Gehirn, in der Regel mit einer Atrophie, meist auch mit Signalauffälligkeiten in verschiedensten Hirnregionen. (Neuroradiology (2006) 48: 613–621) Mit Chelatbildnern kann man die Erkrankung erfolgreich behandeln, manchmal ist eine Lebertransplantation erforderlich. Bei unbehandelten Patienten ist der Verlauf progredient und führt zum Tod. Regelmäßige Behandlung und Überwachung führt meist zu erheblicher Besserung. Die Behandlung ist allerdings lebenslang erforderlich. Unter Behandlung sind Lebenserwartung und -qualität meistens weitgehend normal. Eingesetzt werden D-Penicillamin, Trientine, Zink und Tetrathiomolybdat. Die Behandlung muss ohne Unterbrechung lebenslang fortgeführt werden, auch während einer eventuellen Schwangerschaft. Zu Todesfälle oder schlechtem Verlauf kommt meist bei fehlender Behandlung oder unzureichender Mitarbeit bei der Behandlung.
Quellen / Literatur:
Gute Selbsthilfeseite http://www.morbus-wilson.de/ Wilson’s Disease Association International Wilson’s Disease Support Group UK Morbus Wilson Selbsthilfegruppe Österreich Selbsthilfe Lebertransplantierter Deutschland e.V. Wilson SAK. Progressive lenticular degeneration. A familial nervous disease associated with cirrhosis of the liver. Brain 1912; 34: 295–507.
